2025年1月9日,广州医科大学附属第一医院何建行/刘君/梁文华教授团队联合天津临床多组学重点实验室,在一区期刊CTM(中国科学院/JCR一区,IF= 7.9)在线表发研究 “LcProt: Proteomics-based identification of plasma biomarkers for lung cancer multievent, a multicentre study”。
本研究前瞻性的收集了来自广州和天津的多家中心的287例肺癌患者、肺部良性疾病患者以及102名健康对照的血浆样本,采用基于 NaY 的蛋白质组学技术,实现了高效且具有成本效益的血浆蛋白质组筛查。通过前瞻性队列研究,研究团队发现血浆蛋白质在良性与恶性结节以及不同癌症阶段呈现出动态变化。基于上述发现,本研究构建了多任务、基于人工智能的可解释性预测模型,在肺癌早筛、诊断后淋巴结转移以及 TNM 分期预测方面均展现出更高的准确性。进一步分析表明,在传统临床因素的基础上结合蛋白组学信息,可为肺癌多事件预测提供额外增益。同时,通过基于NaY的蛋白质组学所识别的最小化生物标志物面板,可在保持高诊断和分期准确度的前提下显著降低成本,实现低成本的多任务诊断与分期 (图1,研究摘要图)。
广州医科大学附属第一医院、广州呼吸健康研究院、国家呼吸医学中心何建行教授、刘君教授、梁文华教授为该论文的共同通讯作者;梁恒瑞博士、王润辰硕士、成然硕士为本论文的共同第一作者。

研究摘要图
【研究背景】
肺癌是全球范围内导致癌症相关死亡的首要病因。尽管传统的非侵入性标志物(如 ctDNA、microRNA等)已在肺癌诊疗监测中取得了相当进展,但在准确性、成本效益以及临床验证等方面仍存在局限。相较之下,蛋白质组学因功能变化直接并更稳定,正日益受到学术界的关注,并被视为一种新兴的监测手段。
为克服传统质谱技术在监测深度、高丰度蛋白质掩盖效应等问题2,研究团队联合天津临床多组学重点实验室引入了Zeolite NaY纳米吸附剂与血浆共培养的方法,用于捕获更多种类的蛋白质。研究团队在前期技术创新的基础上开发了基于Zeolite NaY的肺癌多任务预测模型,并进一步探究在肺癌发生发展过程中血浆蛋白动态变化规律和生物学过程。
【研究设计】
1. 采用了前瞻性多中心设计,使用Zeolite NaY纳米材料与血浆共培养的方法,利用LC-MS/MS定量分析了4703种血浆蛋白。
2. 设定了三个主要的任务,包括肺癌诊断(判别肺癌与良性肺部疾病患者/健康对照患者)、淋巴结转移预测(肺癌患者有无淋巴结转移状态)、TNM分期(I-IV期)。
3. 利用UMAP降维、差异蛋白表达(DEP),单蛋白AUC和LASSO回归等方法,逐步筛选不同任务中的关键蛋白。
4. 利用无监督聚类方法(fuzzy)为不同分期对比下的DEPs聚类,揭露肺癌发生发展过程中不同蛋白簇的变化过程。
5. 利用随机森林算法构建多任务预测模型并验证模型性能(AUC),并基于博弈论的解释性人工智能SHAP方法解释血浆蛋白黑箱模型的预测输出。

图2 研究流程示意图
【研究结果】
研究团队定量了4703中DEPs,筛选出与肺癌相关的DEPs(图3)。针对良恶性诊断任务,筛选出300个DEPs,其中255个显著上调,45个显著下调。针对淋巴结转移预测,筛选出920个DEPs,其中716个显著上调,204个显著下调。针对TNM分期预测,在6次比较中筛选出2251个DEPs。
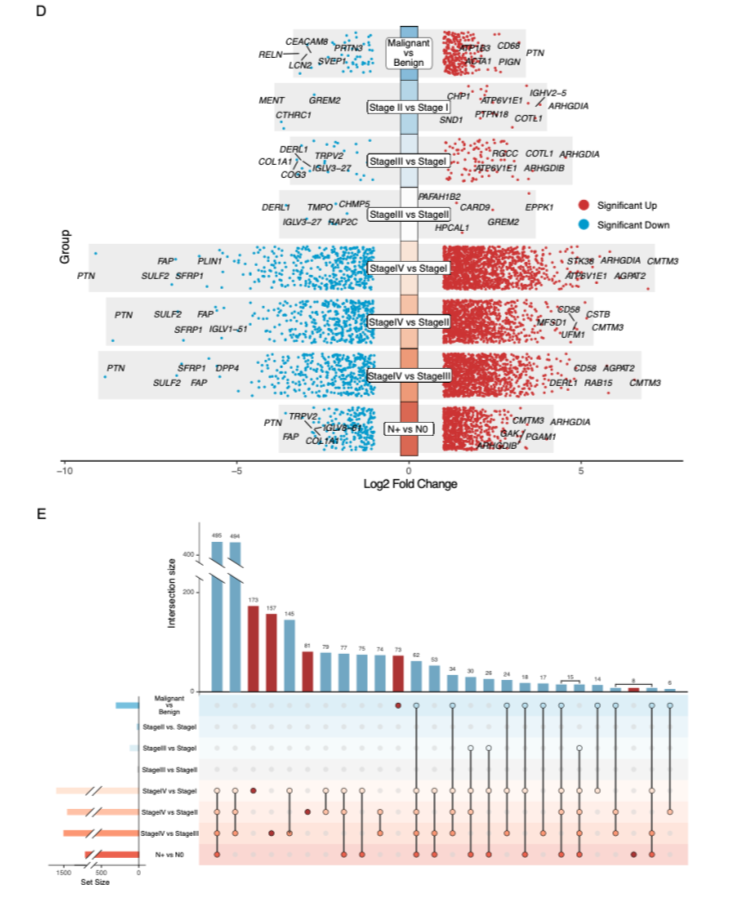
图3 DEP分析结果图
研究团队接下来通过功能富集分析揭示了在不同任务中所表现出的DEPs所涉及的关键生物学事件。针对良恶性诊断任务,细胞膜稳态相关事件、免疫相关相应、细胞应激和凝血相关生物过程显著上调。针对淋巴结转移预测任务,蛋白质运输和细胞因子信号传导、膜稳态相关事件显著上调。
针对TNM预测任务,研究团队利用无监督聚类的方法将差异蛋白划分为6个cluster,并分析了每个聚类在肿瘤不同阶段的动态变化。Cluster1和Cluster 2在肺癌 I-IV 期中呈单调变化。Cluster1的蛋白质随肿瘤进展持续上调,富集在膜稳态和免疫响应通路中。Cluster 2 的蛋白质随肿瘤进展持续下调,与肽翻译和止血相关事件相关。Cluster 3和Cluster 4则代表在晚期(IV期)显著变化 的蛋白质。Cluster3在IV期特异性高表达,与膜稳态和核苷代谢相关。Cluster 4在IV期特异性低表达,与免疫失调和代谢紊乱相关。Cluster 5 和 Cluster 6所代表的蛋白则在肺癌的发生发展过程中出现多次变化。Cluster 5所代表的蛋白质呈现倒U型模式,富集在肽合成和氨基酸代谢通路。Cluster 6所代表蛋白质与膜组织化和能量代谢相关,呈现阶段特异性表达。(图4)
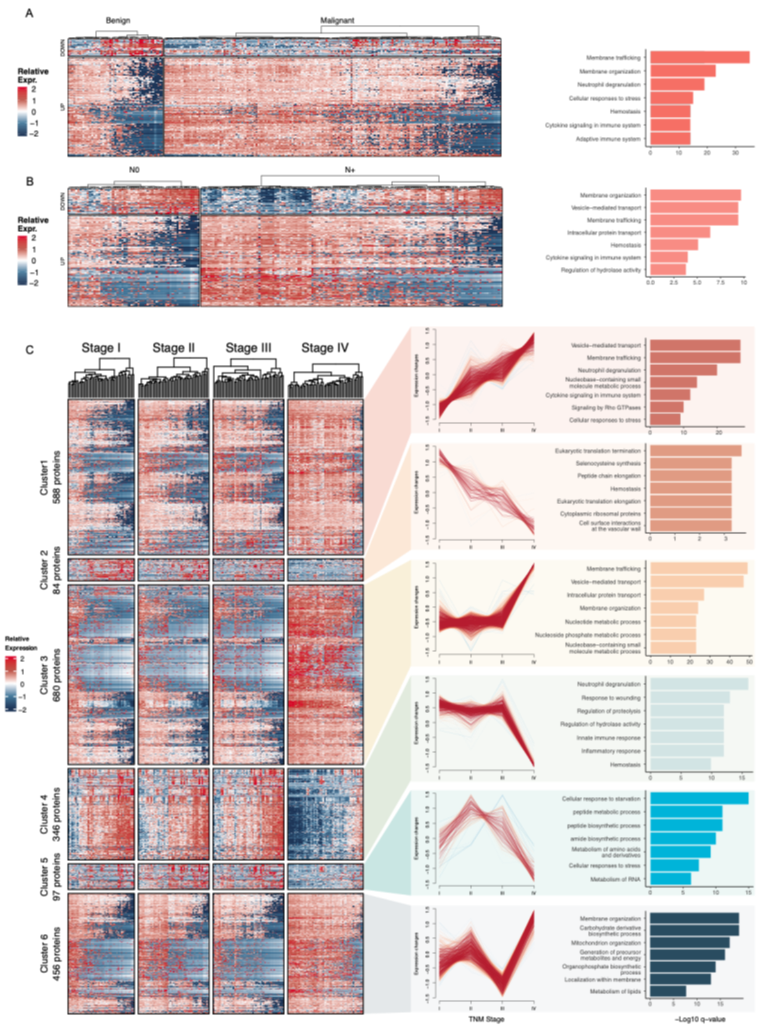
图4 功能富集与蛋白动态变化聚类分析结果
研究团队在筛选DEPs的基础上进一步筛选蛋白后建立了多任务预测模型。对于良恶性诊断任务模型,10蛋白的预测模型在内部验证集中的AUC为0.91,外部验证集AUC为0.97。结合临床信息后,诊断效能进一步提升。针对淋巴结转移预测任务,构建了基于9蛋白的预测模型,模型在内部验证集中的AUC为0.88,结合临床信息后AUC为0.90。针对TNM分期预测任务,构建了10蛋白预测模型,在内部验证集中不同分期中的诊断AUC在0.86至0.99不等,在结合了临床信息后诊断效能均有所提升。(图5)
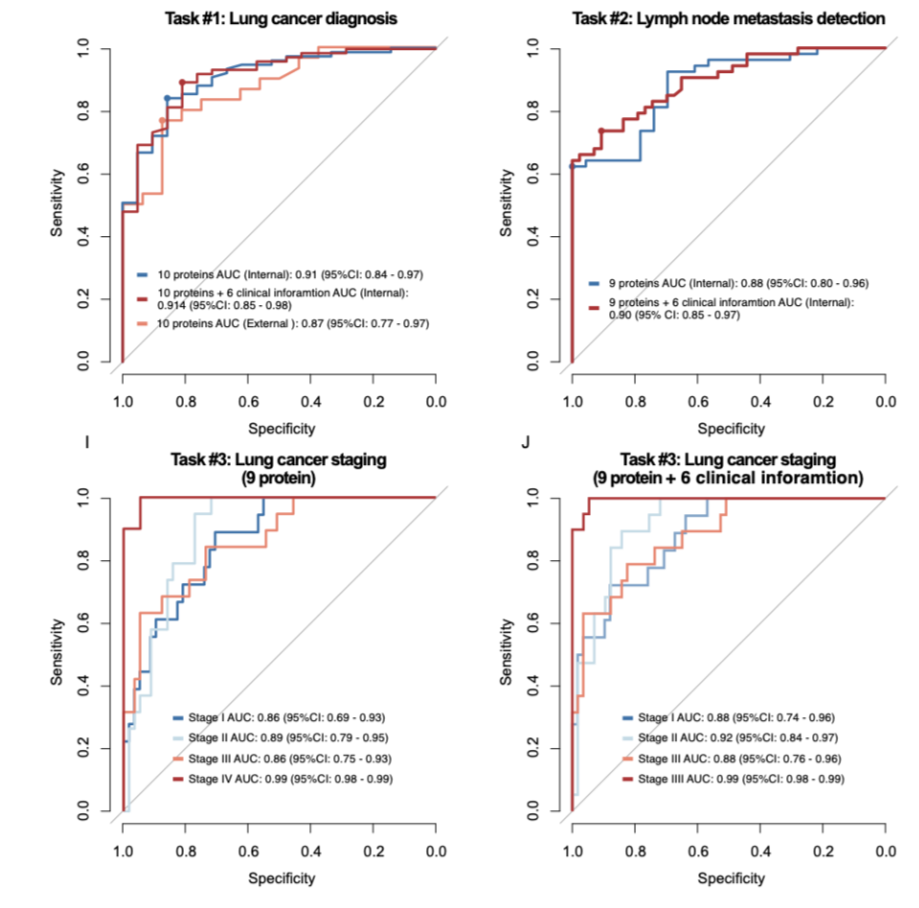
图5 多任务模型诊断效能结果
研究团队接下来利用SHAP算法对随机森林模型进行解释,揭示关键蛋白的风险或保护作用。在良恶性诊断任务中,BAK1、CTSW、PTN是重要蛋白。在淋巴结诊断预测中,XPO1、SND1、NPNT是重要蛋白。而COTL1、PTN、XPO1则在TNM分期预测任务重展现除了分期特异性。(图6)
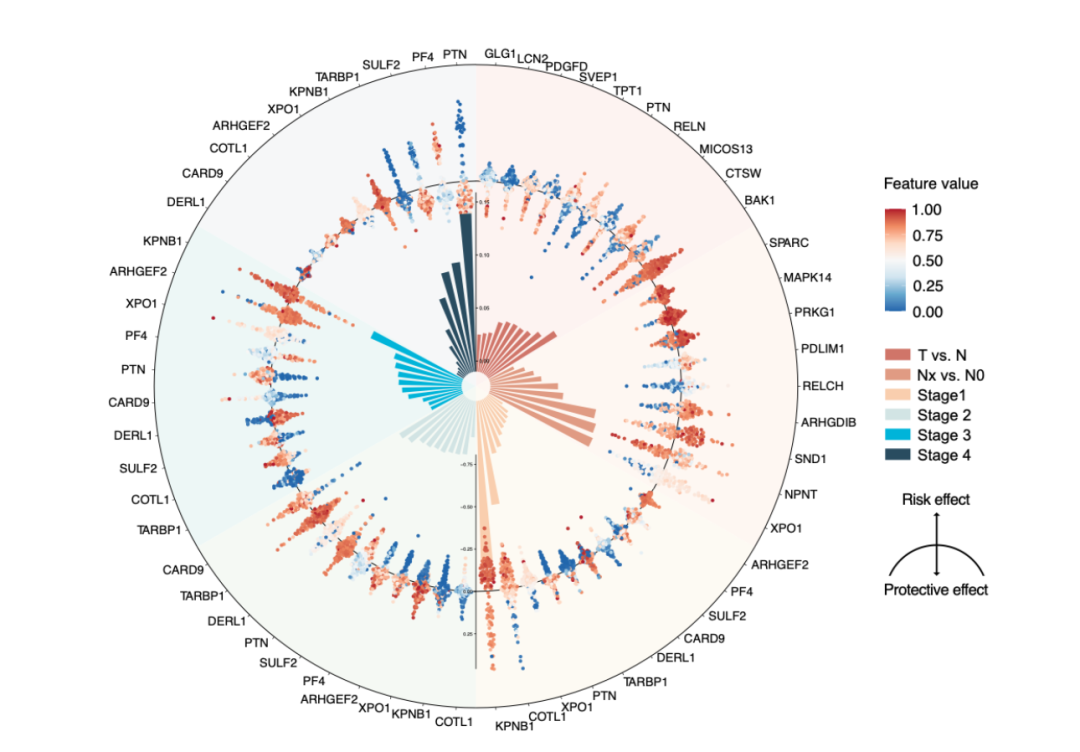
图6 SHAP解释性机器学习结果
最后,研究团队对该良恶性诊断模型在区分肺癌患者与健康对照人群方面的判别能力进行了进一步探究,并采用了 102 名健康对照的血浆样本进行验证。在肺癌与肺良性疾病DEP分析和肺癌与健康对照DEP分析结果的比对中发现,上调蛋白中有 66.78% 的差异蛋白在两组分析中重叠,下调蛋白中有 10.85% 的差异蛋白重叠。肺癌良恶性预测模型的10个蛋白中有9个蛋白在良性肺疾病患者与健康对照者之间表现出一致的表达模式。PCA分析的结果也显示两组人群的蛋白表达谱无明显差异。利用先前发现的10蛋白使用基于10折交叉检验的随机森林方法构建预测模型,AUC大于0.98,说明10蛋白能够良好区分肺癌患者与健康人群。(图7)
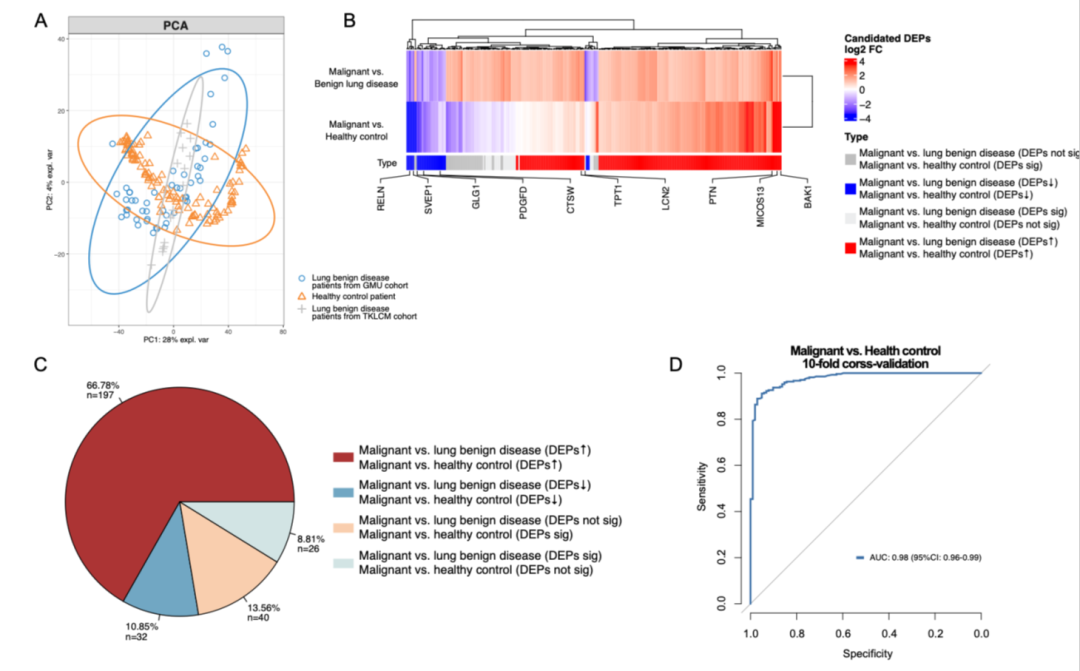
图7 健康人群模型验证结果
参考文献:
1. Liang, H., Wang, R., Cheng, R., Ye, Z., Zhao, N., Zhao, X., Huang, Y., Jiang, Z., Li, W., Zheng, J., et al. (2025). LcProt: Proteomics-based identification of plasma biomarkers for lung cancer multievent, a multicentre study. Clin Transl Med 15, e70160. 10.1002/ctm2.70160.
2. Joshi, A., Rienks, M., Theofilatos, K., and Mayr, M. (2021). Systems biology in cardiovascular disease: a multiomics approach. Nat Rev Cardiol 18, 313-330. 10.1038/s41569-020-00477-1.
 粤公网安备 44010402002638号
粤公网安备 44010402002638号