近日,广州医科大学附属第一医院何建行/梁文华教授团队,在国际期刊MedComm(2023 IF=10.7)在线发表研究“Development of a genome atlas for discriminating benign, preinvasive, and invasive lung nodules”[1]。该研究是迄今为止肺结节/肺癌早检领域最全面的多组学研究,基于全外显子组测序(whole exome sequencing, WES)、转录组测序(RNA-seq)和DNA甲基化测序(DNA methylation sequencing)数据,综合分析了基因突变情况、基因表达情况和基因甲基化水平变化,成功绘制出一份基因组图谱,用于区分良性、浸润前和浸润性肺结节,揭示了相关分子发病机制,并发现了可用于肺癌早期检测的潜在生物标志物和肺癌治疗的药物靶点。广州医科大学附属第一医院、广州呼吸健康研究院、国家呼吸医学中心何建行教授、梁文华教授和基准医疗(AnchorDx)范建兵博士为共同通讯作者;何建行/梁文华团队梁鹏博士、基准医疗彭敏桦博士、陶锦胜、王博以及领星生物魏金旺博士为本文共同第一作者。

【研究背景】
肺癌是全球最常见的恶性肿瘤,早期诊断对于改善患者预后至关重要。然而,低剂量计算机断层扫描(LDCT)筛查肺癌时,常因假阳性结果和误诊而面临挑战。为了解决这一问题,更精准地区分良性肺结节、浸润前肺结节 [含非典型腺瘤样增生(AAH)和原位腺癌(AIS)]与浸润性肺结节[含微浸润性肺腺癌(MIA)和浸润性肺腺癌(IA)],研究团队采集了432例中国肺结节患者施行肺结节切除术所获得的FFPE组织样本进行测序与基因组图谱分析。
【研究设计】
432例肺结节患者FFPE组织样本分为3组,包括良性结节组124例、浸润前肺结节组(AAH+AIS)32例和浸润性肺结节组(MIA+IA)266例,进行了全外显子组测序(whole exome sequencing, WES)、转录组测序(RNA-seq)和DNA甲基化测序(DNA methylation sequencing),再基于测序数据进行了多组学综合分析。
【研究结果】
1. 肺结节体细胞突变谱分析:
● 在浸润性肺结节样本中发现了22个显著突变的基因,其中EGFR(61%)和TP53(24%)是最常见,与既往报道一致
● EGFR突变逐渐增加:良性肺结节,3% → 浸润前肺结节(AAH+AIS),16% → 微浸润肺腺癌(MIA),35% → 浸润性肺腺癌(IA)73%
● TP53突变在浸润性腺癌(IA)中常见(35.43%),在微浸润性腺癌(MIA)中降至2.2%,而在浸润前肺结节和良性肺结节样本中极少被检测到
● 肿瘤突变负荷(TMB)分析显示:入组队列的平均TMB为3.34/Mb,较已报道的晚期肺癌低,其中浸润性肺结节组(MIA+IA)的平均TMB为2.94/Mb;实性结节的TMB显著高于纯磨玻璃结节(p < 0.001);浸润性肺结节的TMB高于良性结节和浸润前肺结节;在IA组,①TMB随着密度增加(纯磨玻璃结节→部分实性结节→实性结节)而逐渐升高,②EGFR突变型亚组(MUT)的TMB中位值显著低于EGFR野生型亚组(WT)(p < 0.01),而这种趋势在良性结节中恰好相反;在纯磨玻璃结节和实性结节中,EGFR突变型亚组的TMB中位值显著低于EGFR野生型亚组(p < 0.001)
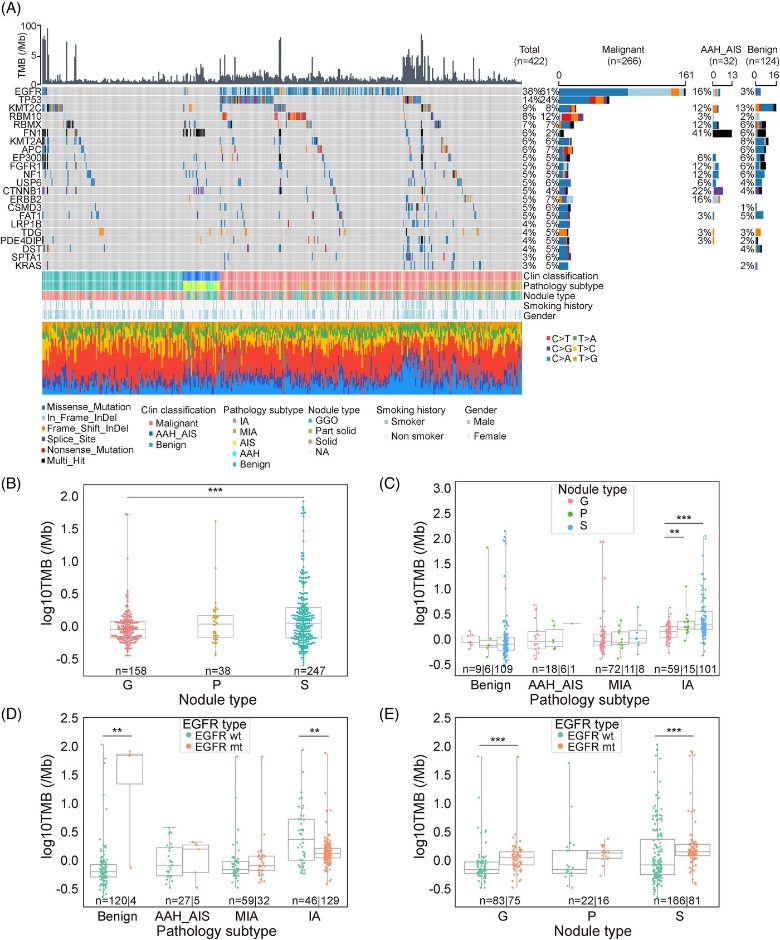
图1. 肺结节体细胞突变全景图
● 转录组测序(RNA-seq)剪切突变分析显示:在浸润性样本中,剪接基因突变导致的变剪切事件数量显著更多;共发现417个基因存在可变剪接事件(剪接事件≥10),通路富集分析显示,主要涉及细胞周期调控和其他癌症相关通路,如KRAS 信号转导和血红素代谢,以RBM10(12%)和RBMX(7%)最为常见

图2. 肺结节基因剪切突变全景图
2. 肺结节转录组特征和免疫微环境分析:
通过对226例肺结节样本(52例良性、29例AAH+AIS、145例MIA+IA)进行转录组测序,结合WES和RNA-Seq数据,基于基因表达谱的无监督层次聚类分析,鉴别出四个具有不同分子特征的亚组:
● 第1组,EGFR驱动组:主要由携带EGFR突变的浸润性肺结节构成,表现为早期和晚期雌激素(ER)和雄激素(AR)应答基因的显著上调
● 第2组,EGFR/TP53共突变组:常见于IA结节组,携带EGFR/TP53共突变,上调了与有丝分裂纺锤体、G2/M检查点及E2F靶点相关的基因,绝大部分基因与细胞周期调控和肿瘤增殖相关,并且糖酵解信号通路也存在上调
● 第3组,炎症组:常见于良性结节及部分MIA/IA肺结节,上调了多种炎症相关基因,如IL-2、IL-6和INF
● 第4组,干细胞样组:主要由浸润前肺结节(AAH+AIS)及部分MIA结节组成,上调了干细胞相关信号通路,包括NOTCH、Wnt/β-catenin和hedgehog信号通路

图3. 肺结节转录组特征和免疫微环境分析
● 研究显示WNT/β-catenin 通路与NK细胞存在强相关性(皮尔逊相关系数PCC=0.8),NOTCH通路与NK细胞存在中等相关性(PCC=0.48);在浸润前肺结节,WNT/β-catenin 通路和NOTCH通路均与NK细胞存在强中等相关性(PCC分别为0.37和0.56)
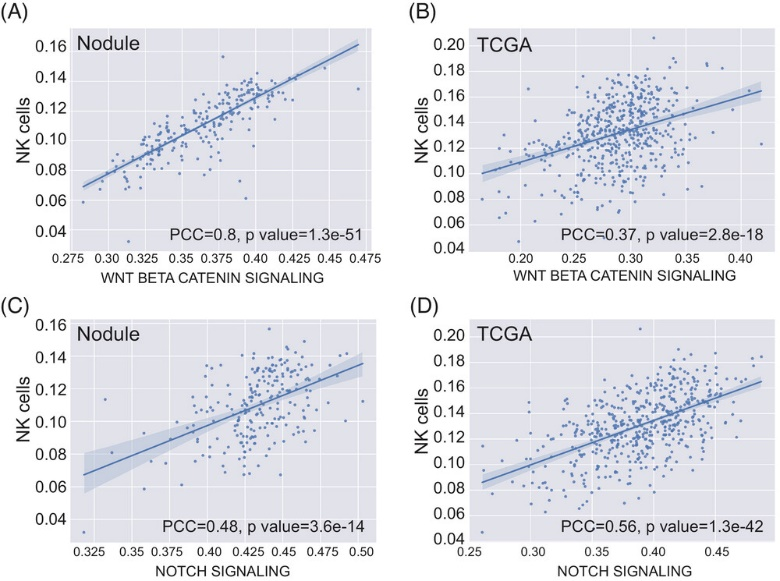
图4. NK细胞与干细胞信号通路之间的相关性分析
3. 肺结节DNA甲基化特征分析:
通过对83例肺结节样本(18例良性、19例MIA、46例IA)进行靶向DNA甲基化测序(基于一个包含12,899个探针位点的定制化甲基化检测Panel),使用无监督层次聚类分析,聚类为M1、M2、M3和M4四组:
● M2组中,分子标志物在良性结节和MIA结节中呈现低甲基化、在IA结节中呈现高甲基化
● M4组中,分子标志物在良性和MIA结节中呈现高甲基化、在IA结节中呈现低甲基化
● 基因通路富集分析显示,M2组中的基因富集于肌肉生成、EMT、IL2/STAT5信号转导及KRAS信号转导的上调;M4组中的基因富集于ER和AR应答通路、TGFβ信号转导以及KRAS信号转导的下调

图5. 肺结节DNA甲基化特征分析
4. 基因表达与DNA甲基化的整合分析:
● 发现了14个基因在甲基化和基因表达方面存在显著的负相关性,这些基因的甲基化改变位于启动子区域
● 在 M2 组中,包括 BDNF、CD48、COX7A1、IKZF1和 TNFRSF1B在内的5个基因的甲基化水平逐渐升高、伴随着基因表达水平的相应下降,从良性结节、MIA结节到IA结节
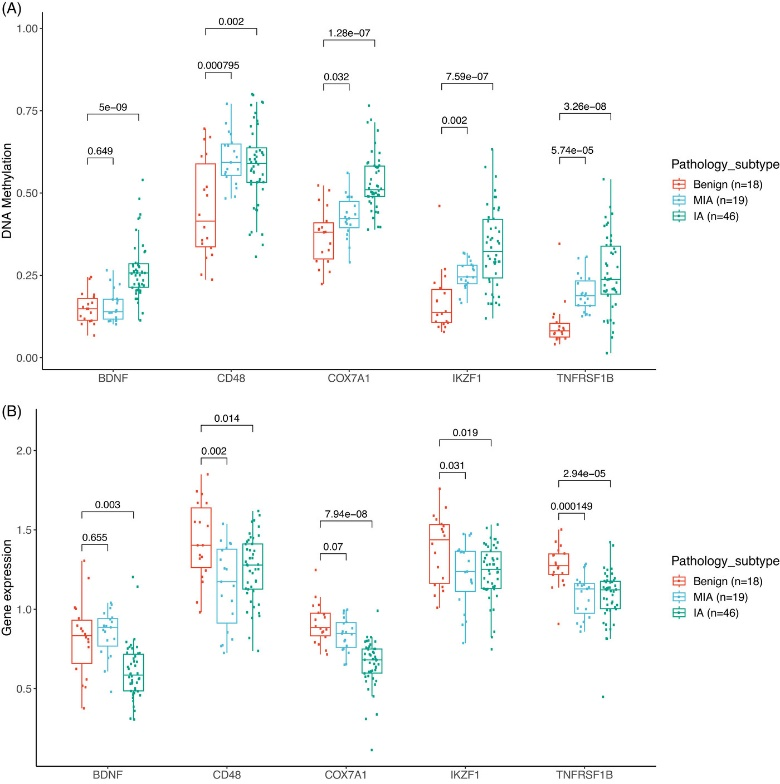
图6. M2组基因表达与DNA甲基化的整合分析
● 在 M4 组中,包括CDC6、CLDN4、EGF、EGFR、ESRP2、GRB7、KRT8、SFN和SLC22A18等9个基因的甲基化水平逐渐降低、伴随着基因表达水平逐渐升高,从良性结节、MIA结节到IA结节

图7. M4组基因表达与DNA甲基化的整合分析
【总结与展望】
本项研究是迄今为止肺结节/肺癌早检领域最全面的多组学研究,旨在阐明中国人群中良性肺结节和早期肺癌的分子发病机制。通过对全外显子组测序、RNA-Seq 和 DNA 甲基化测序的综合分析,绘制出了肺结节的基因组图谱,加深了我们对良性、浸润前和浸润性肺结节不同分子机制的理解。
此外,我们还发现了可用于肺癌早期检测的潜在生物标志物和肺癌治疗的药物靶点。我们的研究为揭示癌症演变和开发阻断解决方案提供了前所未有的资源,具有重大意义。
该研究得到了国家自然科学基金、广州国家实验室专项、广州经济技术开发区创新创业领军人才资助计划、广州市重点研发计划等的资助。
参考文献:
[1] Liang P, Peng M, Tao J, et al. Development of a genome atlas for discriminating benign, preinvasive, and invasive lung nodules. MedComm. 2024; 5:e644. Https://doi.org/10.1002/mco2.644
 粤公网安备 44010402002638号
粤公网安备 44010402002638号